- Sequencing Service
- Bioinformatics Service
- Sample Submission
- Price
- Contact
- Resources
- About
- Careers
- A-Z
- Webmail
- Inside the University Secured Page
-
Search UTHealth Houston

scRNA Immune (Single-cell RNA Immune profiling, 10x Genomics 5’ kit)
Chromium Single Cell Immune Profiling provides a comprehensive approach to simultaneously examine cellular heterogeneity of the immune system, T- and B-cell repertoire diversity, and antigen specificity at single cell resolution. Discover new cell types and states using whole transcriptome analysis, or focus your search with targeted gene expression panels of interest. This service is able to 1) Profile thousands of genes at the single cell level by barcoding mRNA at the 5’ end, for unbiased characterization of cell types and cell states; 2) Simultaneously profile immune repertoire (BCR/TCR) and gene expression from the same cell to enable correlation of clonotype with the corresponding cell subtype; 3) Obtain paired, full-length receptor sequences from T cells and/or B cells with complete isotype resolution, providing functional data for antibody/TCR discovery.
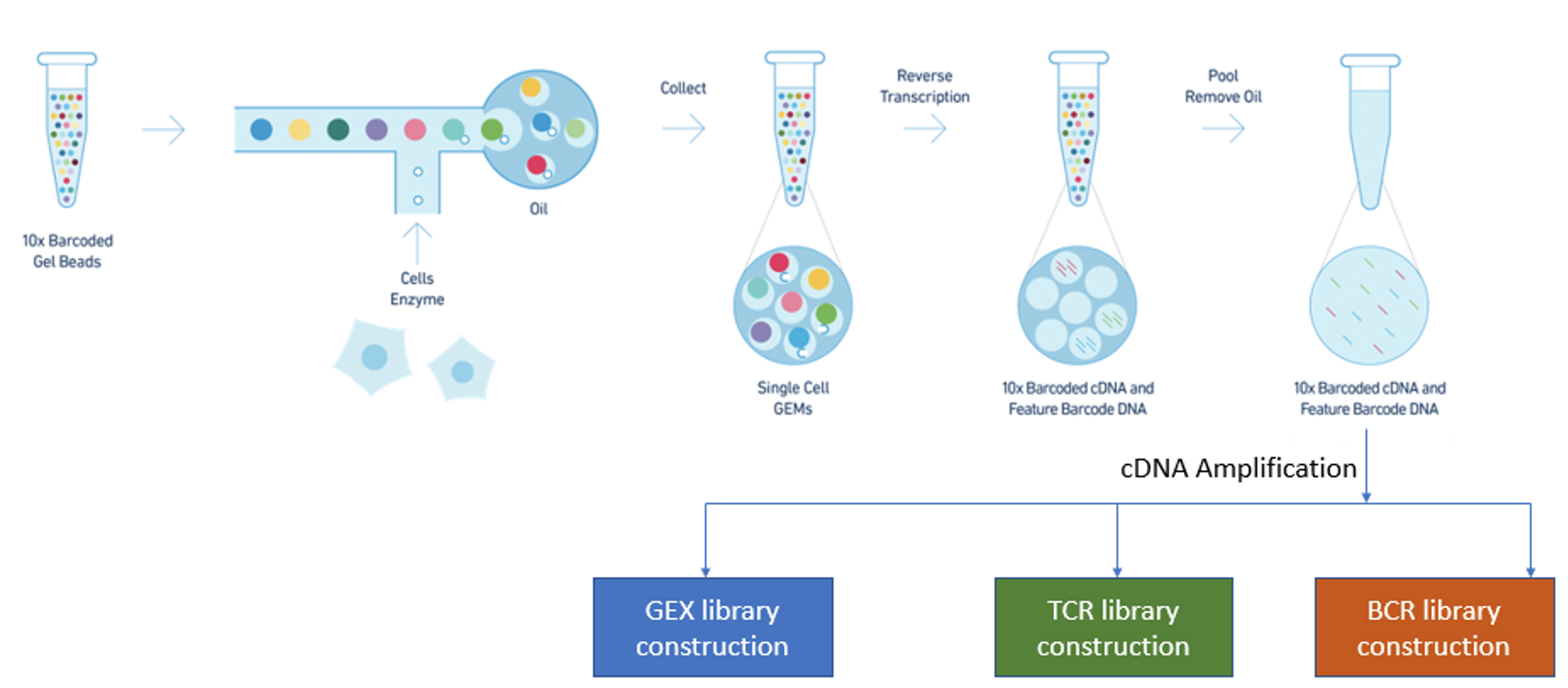
Highlights
Sample Submission requirements
| Service | Sample Type | Minimum Amount |
Minium Cell Viability |
Optimal Concentration |
| scRNA Immune | Singe cells | 50,000 | 85% | 700-1200 cells/ul |
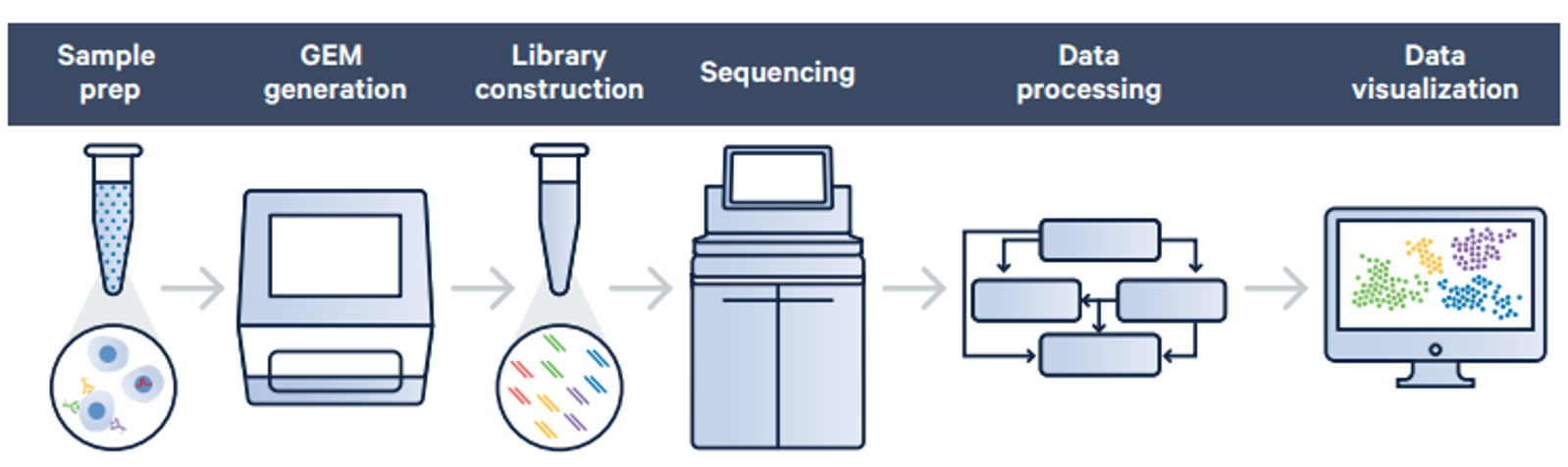
Service workflow
Data analysis with Cell Ranger
cellranger mkfastq demultiplexes raw base call (BCL) files generated by Illumina sequencers into FASTQ files. It is a wrapper around Illumina's bcl2fastq, with additional useful features that are specific to 10x libraries and a simplified sample sheet format.
cellranger vdj takes FASTQ files from cellranger mkfastq or bcl2fastq for V(D)J libraries and performs sequence assembly and paired clonotype calling. It uses the Chromium cellular barcodes and UMIs to assemble V(D)J transcripts per cell. Clonotypes and CDR3 sequences are output as a .vloupe file which can be loaded into Loupe V(D)J Browser.
cellranger count takes FASTQ files from cellranger mkfastq or bcl2fastq for 5' Gene Expression and/or Feature Barcode (cell surface protein or antigen) libraries and performs alignment, filtering, barcode counting, and UMI counting. It uses the Chromium cellular barcodes to generate feature-barcode matrices, determine clusters, and perform gene expression analysis. The cellranger count pipeline outputs a .cloupe file which can be loaded into Loupe Browser for interactive visualization, clustering, and differential expression analysis.
cellranger multi takes FASTQ files from cellranger mkfastq or bcl2fastq for any combination of 5' Gene Expression, Feature Barcode (cell surface protein or antigen) and V(D)J libraries from a single gem-well. It performs alignment, filtering, barcode counting, and UMI counting on the Gene Expression and/or Feature Barcode libraries. It also performs sequence assembly and paired clonotype calling on the V(D)J libraries. Additionally, the cell calls provided by the gene expression data are used to improve the cell calls inferred by the V(D)J library.
cellranger aggr aggregates outputs from multiple runs of cellranger count/vdj/multi. The aggr pipeline normalizes the individual gene expression and feature barcode runs to the same sequencing depth, recomputes the feature-barcode matrices and performs analysis on the combined data. The pipeline also recomputes the V(D)J clonotype grouping on the combined data.
Useful documents